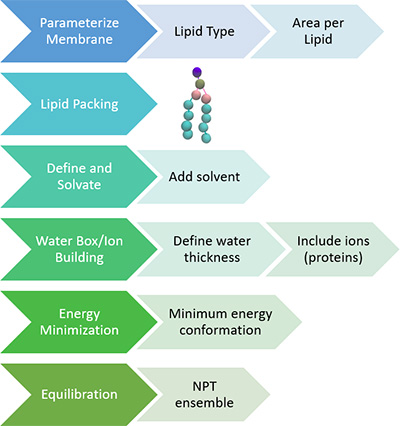
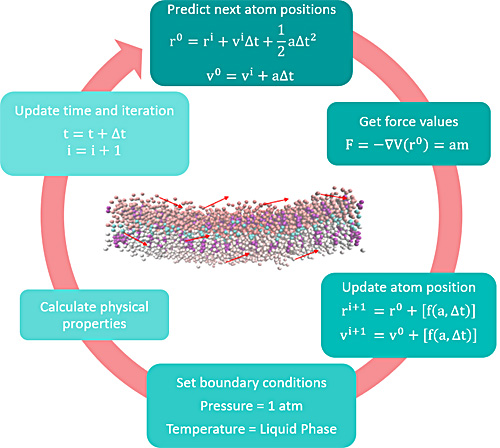
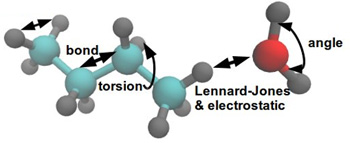
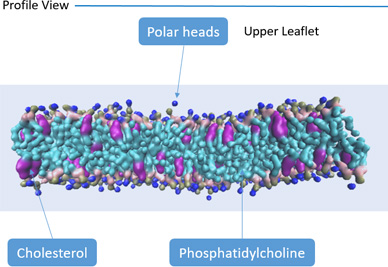
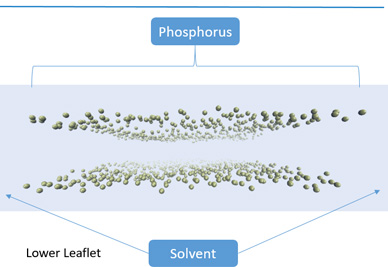
Throughout their life, most types of living cells are subjected to various mechanical forces which play a significant role in development of the cells. Mechanical behavior of the living cells is very important for understanding cells' biological processes such as development, differentiation, disease etc.
In order to investigate elastic and viscoelastic behavior of cells we use Finite Element Analysis (FEA) using software MSC Marc Mentat. In particular, we modeled cells' behavior under loading experienced during two experimental techniques: atomic force microscopy (AFM) and micropipette aspiration.
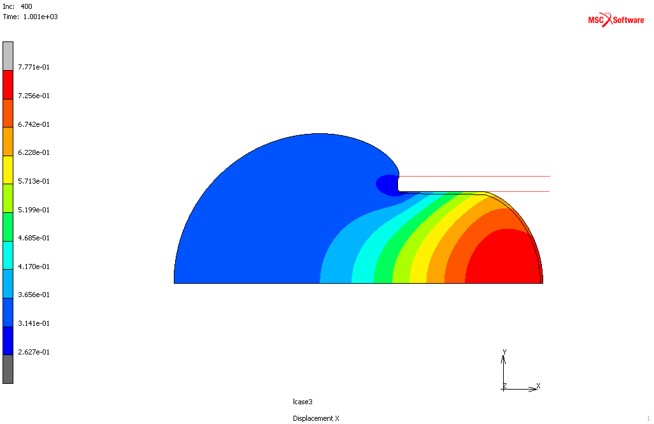
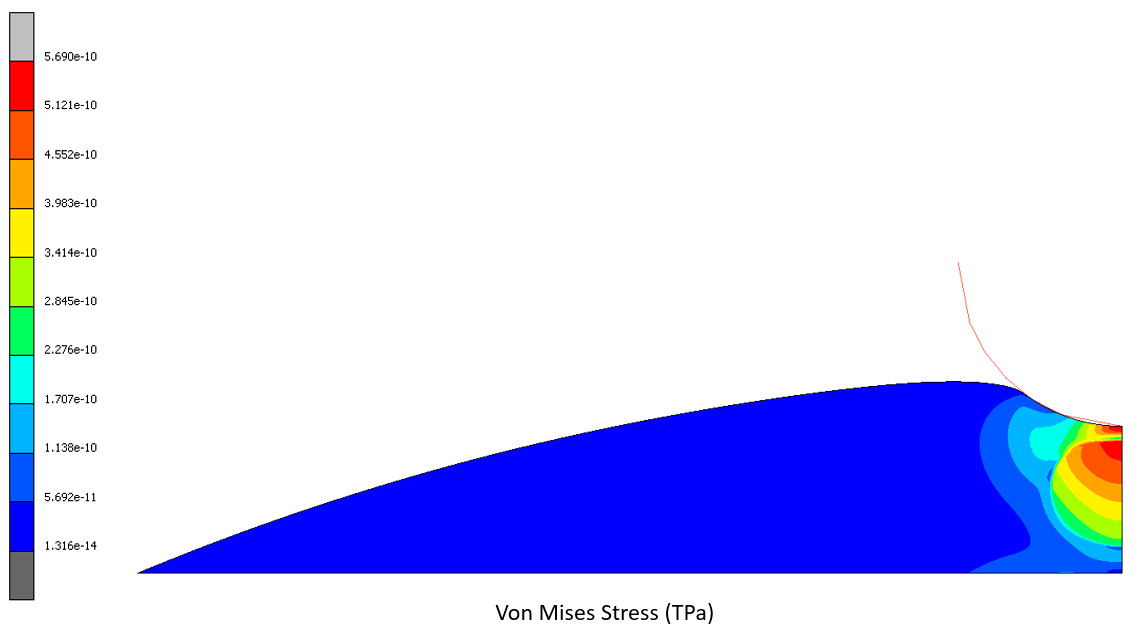
Axisymmetric FEA simulation of nanoindentation of a cell on a substrate
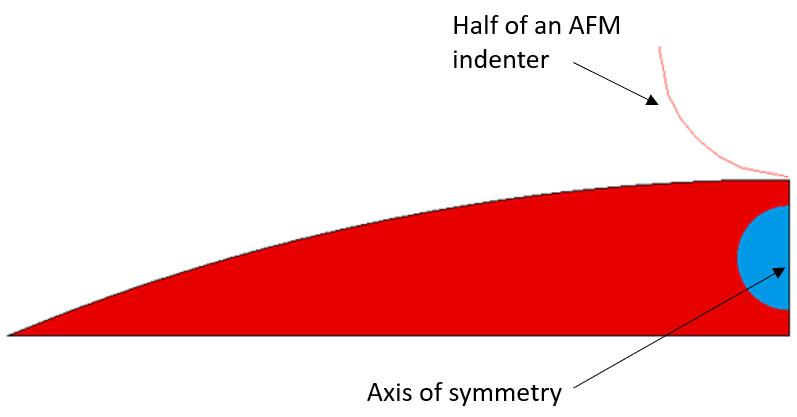
Using atomic force microscopy (AFM) a cell is subjected to indentation on nanoscale and as a result force-displacement curve can be obtained. In this FEA simulation we modeled a cell body (red) and a nucleus (blue). No other internal cell components were modeled. Both cell body and nucleus are described as elastic materials with Young's modulus and Poisson's ratio. The constant force is applied to the center of the cell.
Links to additional images:

Axisymmetric FEA simulation of micropipette aspiration of a cell
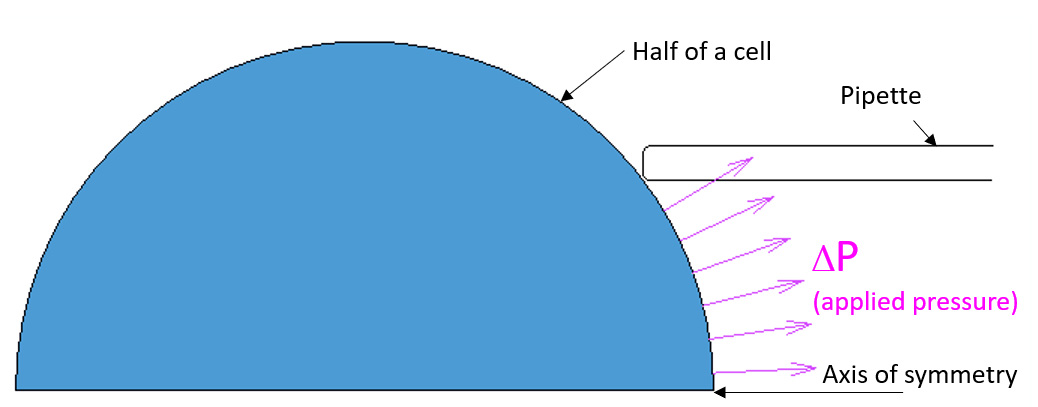
The micropipette aspiration (MA) is a non-invasive experimental technique which is used to study time-dependent deformation of the cells subjected to extracellular pressure ΔP.
In this FE simulation the cell is modeled as aspherical and homogeneous without specifying internal structure. Prony series were used to describe viscoelastic properties of the cell.
Links to additional images: